

Studiare il genoma del virus della Peste Suina Africana (PSA) è come tentare di leggere un manoscritto di migliaia di pagine scritto in un linguaggio sconosciuto, di cui esistono ancora informazioni incomplete. Questo agente patogeno, responsabile di una malattia virale devastante per suini domestici e cinghiali selvatici, rappresenta oggi una delle maggiori minacce per la suinicoltura mondiale. Eppure, nonostante decenni di ricerca, il suo genoma rimane in gran parte ancora da decifrare.
Un virus “fuori scala”
Il primo ostacolo nello studio del virus è la grandezza e complessità del suo genoma. Con dimensioni comprese tra 170 e 193 kilobasi, il virus della PSA possiede il più grande genoma conosciuto tra i virus a DNA che infettano animali. Contiene oltre 150 geni, molti dei quali con funzioni ancora sconosciute o difficili da interpretare.

Per confronto, il virus dell’influenza – ben più studiato – ha solo otto segmenti genomici e poco più di una decina di proteine.
Questa enorme complessità genetica non è solo una curiosità biologica: rappresenta una vera e propria sfida tecnologica. Le regioni ripetute, i geni duplicati e le variazioni strutturali rendono difficile il sequenziamento completo e accurato.
Poche sequenze, molte incognite
A complicare ulteriormente le cose c’è la scarsità di genomi completi condivisi nelle banche dati internazionali. Nonostante la diffusione globale del virus, solo un numero limitato di sequenze è disponibile. Questo limita la possibilità di confrontare i stipiti, comprendere la loro evoluzione e individuare mutazioni chiave legate alla virulenza o alla trasmissibilità.
Le ragioni di questa scarsità sono prevalentemente tecniche e logistiche, ma manca anche in parte una coscienza comune collaborativa tra paesi. La manipolazione del virus della PSA richiede laboratori di alta sicurezza (BSL-3), e solo pochi centri sono attrezzati per eseguire analisi genomiche complete. Inoltre, il sequenziamento di un genoma così grande comporta costi e tempi significativamente più elevati rispetto ad altri virus.
Il Caso Italiano: un Laboratorio Naturale
Un contributo fondamentale alla conoscenza del virus della PSA è arrivato recentemente dall’Italia. Lo studio “Molecular Characterization of the First African Swine Fever Virus Genotype II Strains Identified from Mainland Italy” (Giammarioli et al., 2023) ha fornito la prima caratterizzazione molecolare dei ceppi di genotipo II identificati nella penisola, rivelando la stretta parentela con quelli diffusi in Europa orientale.
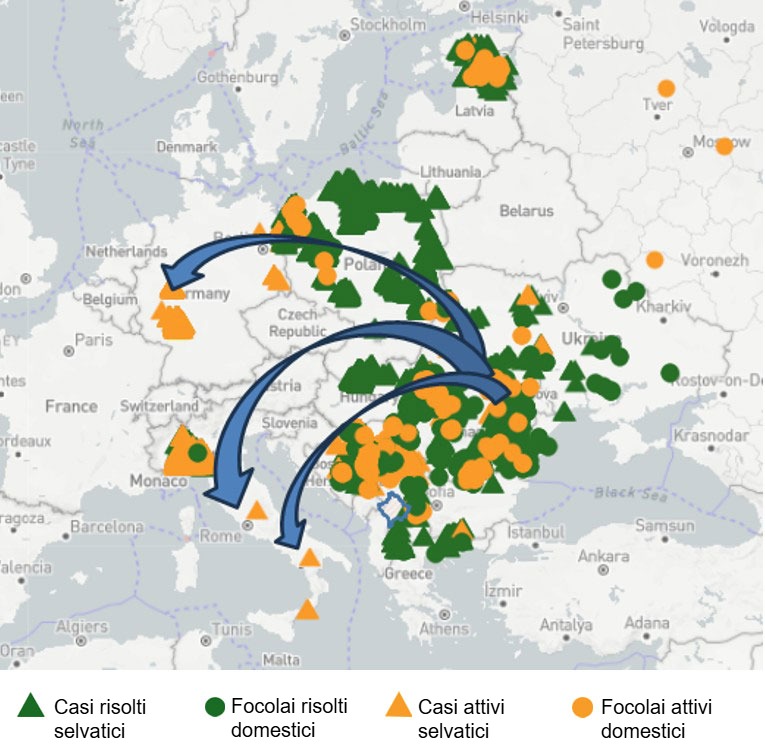
Successivamente, un altro lavoro pubblicato nel 2025, “Genome-Wide Approach Identifies Natural Large-Fragment Deletion in ASFV Strains Circulating in Italy During 2023” (Torresi et al., 2025), ha identificato delezioni naturali di grandi dimensioni in alcuni ceppi stipiti. Tali mutazioni, che comportano la perdita di intere porzioni di genoma, potrebbero influenzare la virulenza o la capacità del virus di eludere la risposta immunitaria, ma il loro significato biologico resta da chiarire.
Genotipi, sierotipi e la diffusione globale
Il virus della PSA è classificato in almeno 24 genotipi distinti, basati sulla sequenza del gene B646L (che codifica per la proteina p72), e quasi altrettanti sierotipi, 8 i più comuni, determinati dalla variabilità della proteina CD2v (EP402R). Tuttavia, la correlazione tra genotipo, sierotipo e virulenza non è ancora ben compresa: virus appartenenti allo stesso genotipo possono avere comportamenti epidemiologici diversi.
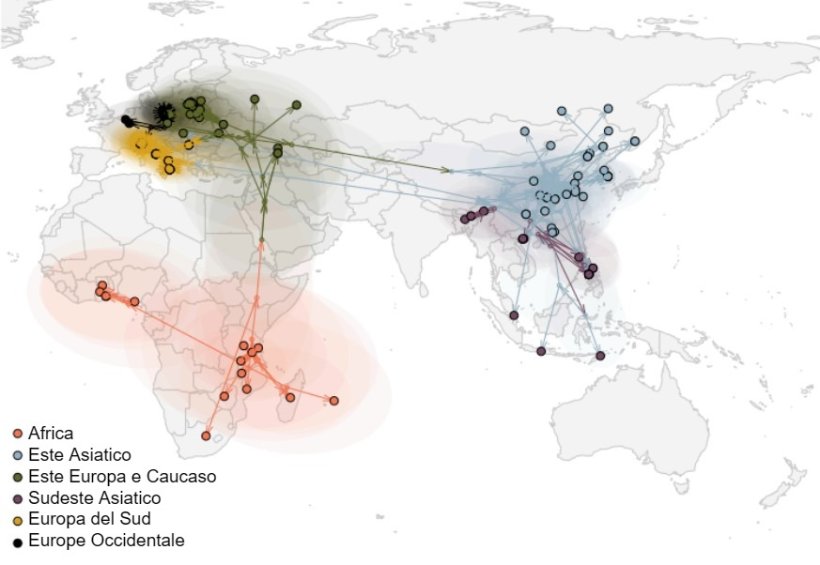
Un recente studio filogenetico, “A phylogenetic contribution to understanding the panzootic spread of African swine fever: from the global to the local scale” (Rossi et al., 2025), ha contribuito in modo decisivo a chiarire i meccanismi di diffusione del virus su scala globale e locale, evidenziando come piccoli eventi evolutivi e mutazioni puntiformi possano influenzare la geografia delle epidemie. Questo tipo di approccio filogenetico è cruciale per tracciare la “storia filogeografica” del virus ed interpretare le sue rotte di diffusione.
Nuove tecnologie, un puzzle da ricostruire
Oggi, le nuove piattaforme di sequenziamento e gli strumenti di bioinformatica avanzata stanno aprendo prospettive inedite. Le tecniche di long-read sequencing permettono di leggere tratti di DNA molto lunghi, riducendo gli errori nelle regioni ripetute. Parallelamente, l’integrazione con dati proteomici e strutturali potrebbe aiutare a decifrare la funzione dei molti geni ancora “orfani di significato”.
Tuttavia, finché i genomi completi disponibili resteranno pochi e la cooperazione scientifica internazionale limitata, il virus della Peste Suina Africana continuerà a essere, geneticamente parlando, un gigante avvolto nel mistero.

