

Il virus dell'influenza A (IAV) è uno dei patogeni respiratori più importanti in suinicoltura. La malattia ha un impatto sulla mortalità e cause perdite economiche importanti causate dalla minor produttività e costi associati alla vaccinazioni e trattamenti. Inoltre, dovuto alla sensibilità dei suini ai virus influenzali di varie specie, in particolare dall'uomo, possono emergere nuovi virus a causa del riordino genetico, con ripercussioni sulla salute pubblica. Di conseguenza, la comprensione della diversità genetica dei virus circolanti nei suini permette l'identificazione di nuovi linee virali, offrendo un criterio su cui basarsi e su cui migliorare le strategie di intervento.
La diversità genetica dell' IAV suino negli USA è prodotto dalla derivazione e dal cambio antigenico, assieme all'introduzione di IAV di altre specie nelle popolazioni suine (Vincent et al., 2008). In termini generali, esiste una co-circolazione di 3 subtipi dominanti (H1N1, H1N2 e H3N2) e 4 linee genetiche principali descritte secondo l'origine dei tratti genici. La classica linea suina H1 ha origine nella influenza spagnola pandemica nel 1918. La seconda linea fu rivelata alla fine degli anni 90 a partire dall'emaglutinina (HA) e dalla neuraminidasi (NA) derivate dall'influenza stagionale H3N2. Questa linea era un virus nuovo riordinato che conteneva tratti genetici HA, NA e PB1 derivati dall'influenza stagionale dell'uomo H3N2, tratti genici PB2 e PA dell'influenza aviare e segmenti genici NP, M e NS dell'influenza suina classica H1N1, per questo denominato virus "riordinato triplice-triple reassortant". Questi virus si riordinarono a loro volta con virus classici H1N1 per acquisire la HA e/o la NA, portando a nuovi tipi genetici di virus H1N1 e H1N2. La costellazione di geni interni del triplice riordinamento (TRIG) è rimasto relativamente consistente con quelli di origine suina (geni M, NP e NS), aviare (geni PB2 e PA) ed umani (PB1) dei virus dell'influenza. La terza linea si è creata a partire da ripetute infezioni del virus stagionale umano H1 IAV nei suini (quello che si conosce come "spillover", o, l'evento nel quale un patogeno tipico di una specie, salta ad un'altra specie) nel principio del secolo XXI, creando due linee distinte di virus stagionali nell'uomo H1N1 e H1N2. La quarta e più recente linea, rappresenta un "spillover" del virus umano stagionale H3N2 che ha avuto luogo nel 2010-11. In questo scenario 4 linee principali di HA: il virus continua ad evolversi, formando nuovi clusters genetici (figura 1).

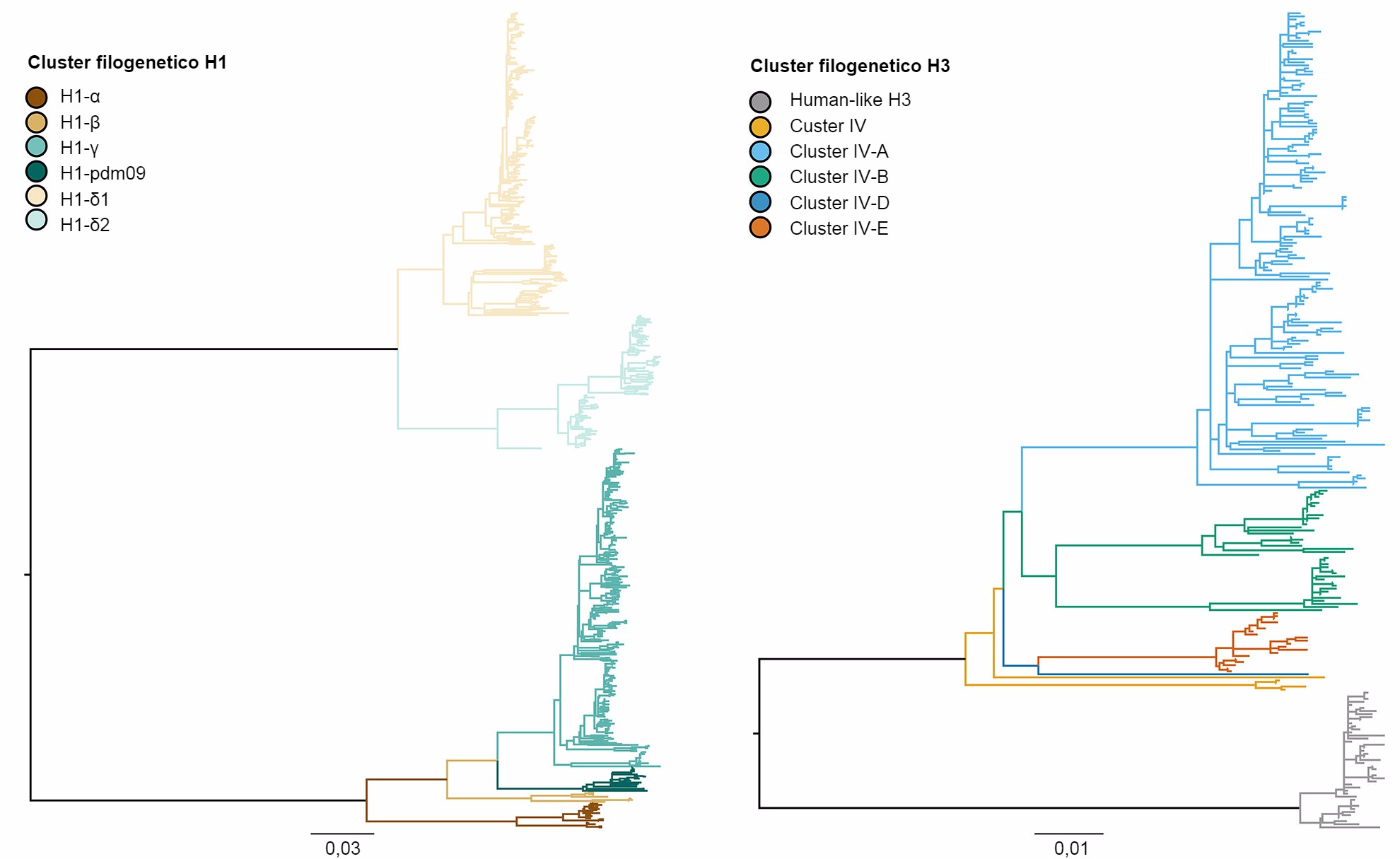
Figura 1. Albero filogenetico che descrive le correlazioni genetiche tra sequenze geniche della emaglutinina dell'influenza A suina H1 e H3 del 2015 generate usando metodi di massima verosimilitudine. Le ramificazioni in colore rappresentano le diverse categorie di cluster. Le lunghezze dei rami sono disegnate in scala e la barra della scala indica il numero di sostituzioni nucleotidiche per sito.
In uno sforzo per migliorare la compresione della diversità genetica dell'IAV suino e monitorare il virus pandemico 2009 H1N1 (H1N1pdm09) nei suini, nel 2009 è stato posto in pratica un sistema nazionale di vigilanza da parte del Ministero dell'Agricoltura degli USA (USDA) con il sostegno dei laboratori di salute animale a livello nazionale (NAHLN). Il sistema si basa sugli invii anonimi da parte di allevatori e veterinari; tuttavia, il sistema manca di informazione geografica sulla provenienza dei campioni. I dati epidemiologici raccolti includono lo stato, il tipo di campione, il motivo dell'invio, l'età, la localizzazione, risultati del test e, se applicabile, il sequenziamento dei geni HA, NA e M che vengono depositati in base ai dati on line delle sequenze del Centro Nazionale dell'Informazione Biotecnologica GenBank (Korslund, et al. 2012; Anderson, et al. 2013). . Questi dati assieme all'investimento continuato nel sistema, hanno prodotto informazioni sui modelli di diffusione dell' IAV, sulla diversità genetica lungo l'anno e sulla dinamica dell'evolulzione dell' IAV nel NordAmerica dal 2010 fino ad oggi (Anderson, et al., 2013; Anderson, et al., 2015; Lewis, et al., 2014; Rajão, et al., 2015)..
I 3 subtipi (H1N1, H1N2 e H3N2) endemici nella popolazione suinicola negli USA sono stati identificati nella popolazione americana dal 2010 fino ad oggi. I subtipi H1N1 e H1N2 sono stati identificati con frequenze similari (~35%) duranti i 5 anni. Il subtipo H3N2 rappresenta ~30% dei virus identificati in tutto il periodo. Per scoprire la diversità genetica dei virus H1 si utilizza un sistema basato in lettere dell'alfabetto greco per dividere i dati dei clusters di HA all'interno delle linee classiche (α, β, γ, γ-2 y H1N1pdm09) o della linea umana (δ-1, δ-2). Analogamente, i geni del cluster IV di H3 si separano in 6 clusters genetici(A a F) e un clusater H3 tipo umano (Rajão, et al., 2015; Kitikoon, et al., 2013).. Indubbiamente, nonostante il potenziale di circolazione dei 14 clusters genetici, la maggior diversità delle HA osservata negli allevamenti suinicoli americani si limita a 3 clusters. Da gennaio 2015 a dicembre 2015, il 43% dei ceppi isolati H1 appartenevano al cluster γ della linea classica e il 37% si classificarono come δ-1 della linea stagionale umana; il 20% rimanenti dei virus H1 provenivano dai clusters δ-2, α, H1N1pdm09 e β. Degli 8 clusters potenziali H3, il cluster IV-A rappresenta la maggioranza di IAV H3 circolanti nei suini e negli allevamenti commerciali (il 61% dei virus H3 durante 2015), furono identificati in modo crescente i ceppi isolati come H3 tipo humano (dal 5% a dicembre 2014 al 25% a dicembre 2015), mentre il resto dei ceppi isolati rappresentano identificazioni sporadiche dei clusters IV-B, IV-C, IV-D, IV-E.
Per quanto riguarda l'efficacia dei programmi vaccinali, esiste una rinnovata preoccupazione rispetto alla neuraminidasi (NA)che possa svolgere un ruolo importante a causa della sua crescente diversità (Sandbulte et al., 2016). Tutto questo complica la situazione, la NA ha molte meno linee rispetto all'HA. La HA è parallela con i geni N2 derivati da uno o due linee umane stagionali di H3N2 (sia del 1998 sia del 2002: (Nelson et al., 2011)), geni N1 della linea classica suina, o geni della linea stagionale umana H1N1 (Anderson et al., 2013). L'attuale IAV suino circolante, l' N1 appartiene, alla linea classica (94%) e l' N2 alla linea 2002 (83%), mentre la linea 1998 rappresenta, anche se non rilevato in modo consistente, un piccolo componente dei ceppi isolati di IAV.
Una seconda preoccupazione è correlata con i modelli spaziali nella diversità dell'IAV suino. Gli Stati Uniti sono suddivisi in 5 regioni, che riportano i casi con base nei distretti veterinari stabiliti dall' USDA-APHIS (figura 2A). Ci sono sottili ed importanti differenze tra le regioni quanto alla diversità e abbondanza di campioni presenti nel sistema nazionale di vigilanza dell'USDA (figura 2B). Mentre nelle regioni 1 e 2 sono più presenti H1N1, la regione 3 presenta più H1N2 e la regione 4 più H3N2. Considerando che la popolazione suinicola è di 1,6 milioni di suini (USDA-NASS, 2012), ci sono troppo pochi dati per la Regione 5. La regione 2 mostra la maggior diversità in termini di differenze di clusters HA e NA osservati e la maggior parte dei ceppi isolati di IAV suini provengono da questa regione. In questo modo, la distribuzione dei clusters HA e NA suggerisce che le decisioni regionali o locali dei componenti vaccinali sono di grande importanza.
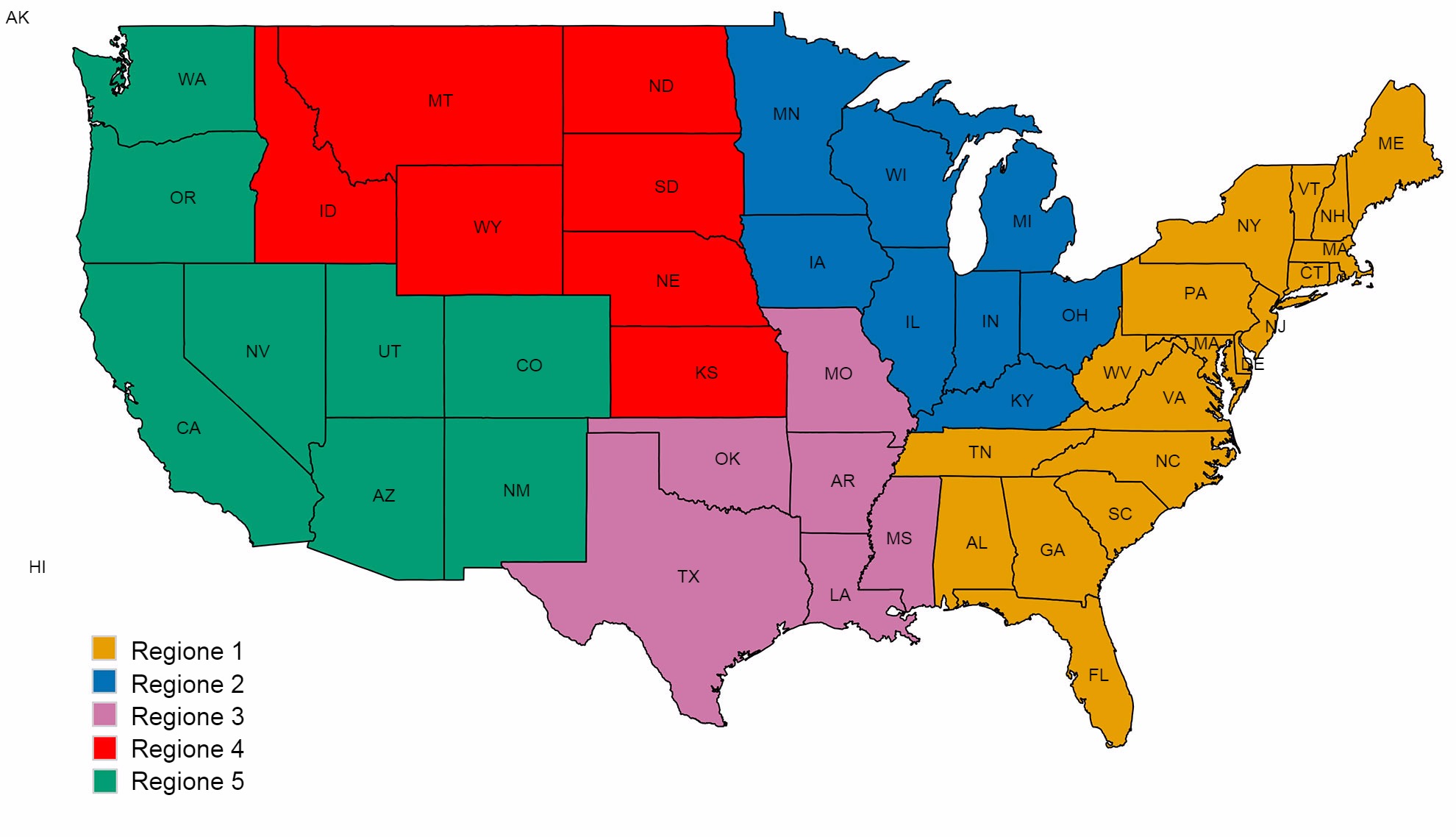
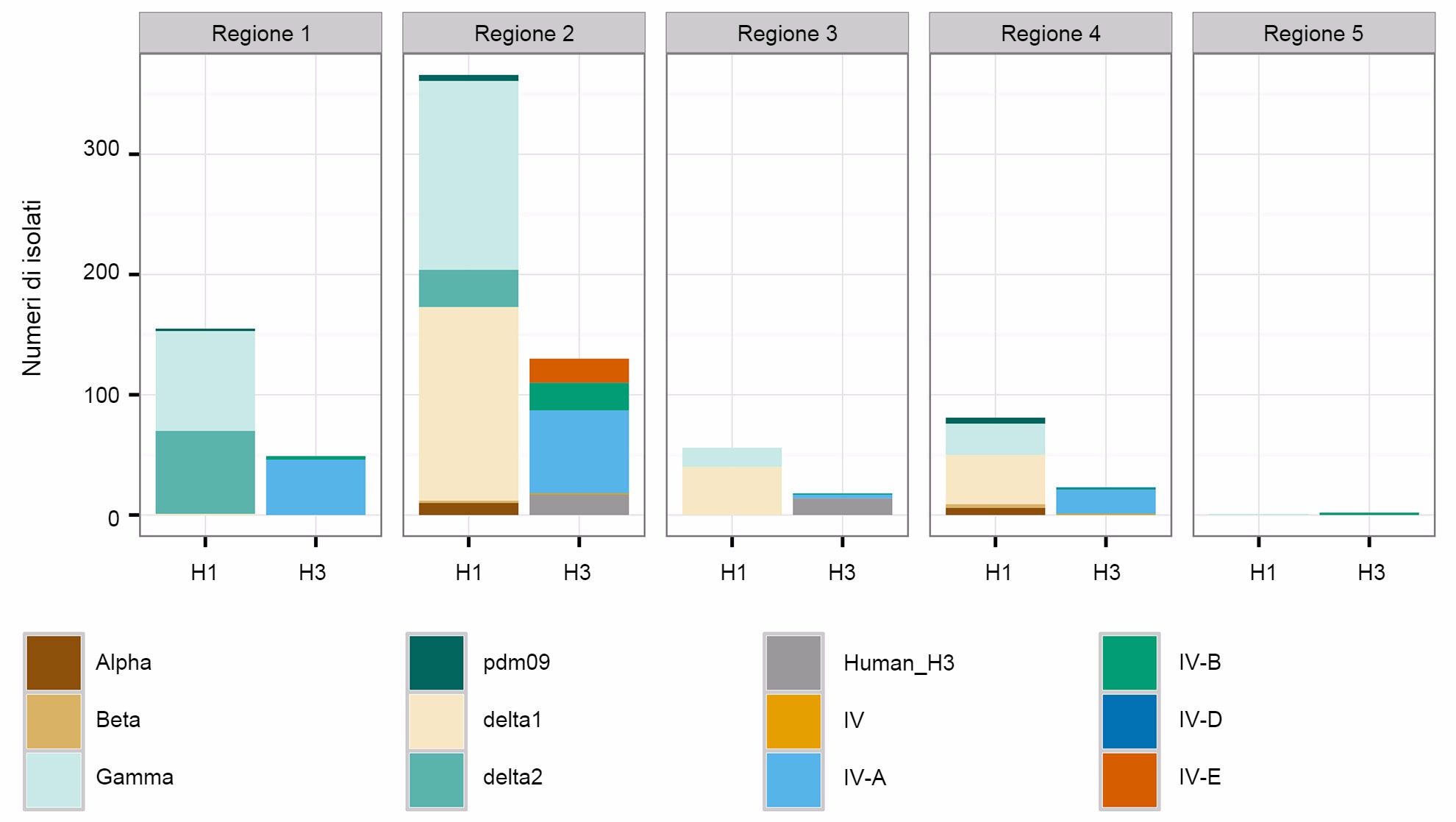
Figura 2. Regioni veterinarie dell'USDA-APHIS (A). N° di isolati di influenza suina A raccolti in ogni regione durante il 2015 e classificati secondo il claster filogenetico e colorato come nella figura 1 (B).
La diversità genetica dell'IAV suino è un tema complesso a livello regionale e specialmente a livello globale. Negli USA, ci sono 17 clusters genetici che sono emersi e persistono dopo gli eventi di "spillover" tra ospiti non suini (ossia, uomini) e i conseguenti processi ecologico-evolutivi. Nonostante sia formata di clusters genetici diversi di vari episodi di "spillover" nei suini, la diversità genetica somiglia a quella della popolazione mondiale (per esempio Watson, et al.,2015; Bahl, et al.,2015; Vijaykrishna, et al. 2015). La genetica dell' IAV suina moderna può essere usata per realizzare studi sull'evoluzione e diversità antigenica: questi studi dovrebbero essere portati a termini con i dati attuali disponibili dalle popolazioni regionali. Questo assieme di dati, con l'implementazione di un programma vaccinale adeguato, offrirebbe informazioni fondamentali per l'uso nella selezione di ceppi vaccinali e per l'implementazione delle politiche di gestione della sicurezza della salute animale e pubblica.

