

Il virus della sindrome riproduttiva e respiratoria suina (PRRSV) è, insieme al virus della peste suina africana, uno dei patogeni con il maggiore impatto economico sul settore suinicolo globale. La recente comparsa di ceppi di PRRSV altamente aggressivi, come il PRRSV altamente patogeno (HP-PRRSV) in Asia, il Rosalía in Europa e il L1C.5 in Nord America, ha acceso il dibattito sulla necessità di continuare a migliorare la diagnosi e il controllo del PRRSV.
L'impiego della RT-PCR per rilevare il materiale genetico del PRRSV è di routine in tutto il mondo per effettuare lo screening del virus nelle popolazioni.

Un passo avanti rispetto al rilevamento dell'RNA è il sequenziamento genetico di tutto il PRRSV, che viene solitamente eseguito utilizzando la tecnica Sanger. In tutto il mondo, la porzione del virus più comunemente utilizzata per il sequenziamento del PRRSV è l'ORF5, sebbene alcuni laboratori generino report con il sequenziamento di ORF7.
L'ORF5 rappresenta circa il 4% e l'ORF7 il 2,4% del genoma del vPRRS, quindi non forniscono una copertura genetica completa dell'intero genoma.
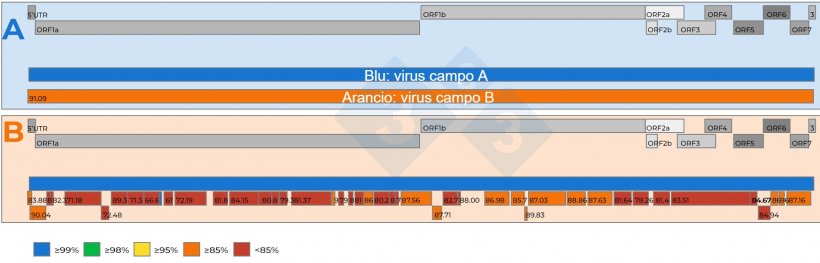
Negli ultimi anni è cresciuto l'interesse per l'uso del sequenziamento di nuova generazione (NGS-next-generation sequencing) per il recupero di genomi completi del PRRSV a fini di indagini epidemiologiche all'interno di un'allevamento o di un sistema di produzione (Figura 1).
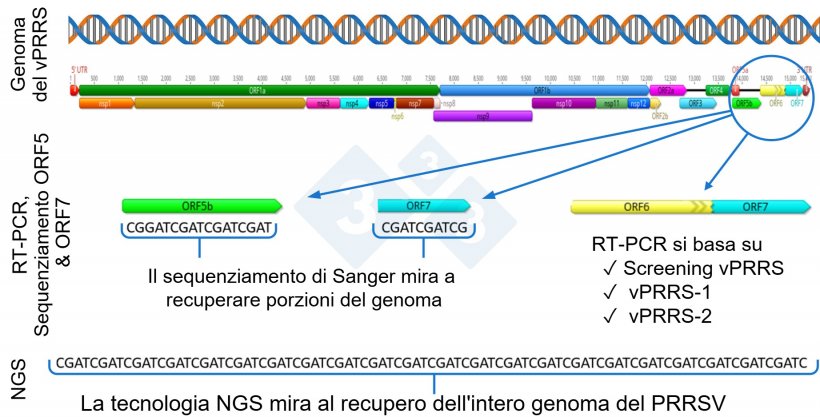
Durante il processo di replicazione, il PRRSV subisce cambiamenti e mutazioni genetiche, che possono potenzialmente portare alla comparsa di nuove varianti virali. È noto che il PRRSV ha un elevato tasso di mutazione, pari a circa lo 0,5-1% all'anno, distribuito in modo non uniforme tra le diverse regioni del genoma, i diversi tipi di virus e i diversi lineages genetici, il che porta ad una costante evoluzione genetica. In particolare, la mutazione e l'evoluzione genetica del virus possono verificarsi in tutti i geni, quindi il sequenziamento di una sola parte del genoma, ad esempio ORF5 o ORF7, rischia di non rilevare i cambiamenti che si verificano al di fuori della regione sequenziata (Figura 1).
In questo scenario, la tecnologia NGS diventa uno strumento utile in quanto offre l'opportunità di recuperare un genoma completo del PRRSV da utilizzare nelle indagini epidemiologiche.
In che modo veterinari ed allevatori possono trarre vantaggio dal NGS?
Per massimizzare l'utilità dell'NGS, è opportuno considerare alcuni punti:
- È importante che il veterinario e il laboratorio diagnostico siano coinvolti fin dall'inizio per allineare la diagnosi differenziale, le aspettative e l'approccio ai test.
- Disponiamo di una sequenza completa del genoma di un ceppo di riferimento del PRRSV proveniente dall'allevamento o dal sistema di produzione per il confronto?
- In caso contrario, utilizzare la tecnica NGS su campioni PRRSV RT-PCR-positivi con bassi valori di Ct, idealmente <24, per recuperare un genoma completo da utilizzare come ceppo di riferimento per l'allevamento. Il valore di Ct è inversamente proporzionale al numero di copie genetiche virali presenti nel campione; ovvero, più basso è il Ct, maggiore è la carica virale attesa e maggiore è il successo della tecnica NGS;
- Un ceppo di riferimento consente il successivo confronto dei genomi recuperati in modo prospettico per comprendere l'evoluzione genetica del vPRRS all'interno dell'allevamento, del flusso e del sistema.
- Qual è lo scopo del realizzare un NGS?
- Se l'obiettivo è rilevare l'evoluzione del virus a livello dell'intero genoma, è opportuno prelevare campioni di polmoni e di siero, poiché è più probabile che questi campioni consentano di recuperare un genoma completo.
- Se l'obiettivo è comprendere la diversità virale in un'allevamento, è consigliabile utilizzare tipologie di campioni di popolazione come gli emosieri (processing fluid), fluidi orali o campioni individuali in pool. Ad esempio, i campioni di siero aggregati hanno maggiori probabilità di recuperare virus multipli se presenti nel campione.
- Le infezioni miste con due o più virus PRRS in un allevamento sono una realtà (Figura 2a) e se i due virus sono molto simili, l'NGS potrebbe non recuperare un genoma completo;
- Se nel campione sono presenti più virus, l'NGS può recuperare frammenti del genoma, noti come "contig", da virus diversi che, confrontati con un ceppo di riferimento dell'allevamento, possono aiutare a discernere se nel campione sono presenti più virus (Figura 2b);
- Infine, assicurati di avere qualcuno che ti aiuti con le analisi genetiche e i confronti, e che coordini i tempi di consegna, poiché l'NGS è spesso un processo lento che può richiedere più di una settimana per essere completato.
Che tipo di informazioni epidemiologiche possiamo ottenere dall'NGS?
I risultati generati da NGS possono aiutare a rispondere a domande come:
- Il virus si è evoluto attraverso sostituzioni nucleotidiche casuali nelle stesse posizioni genomiche? Quanto è cambiato il virus tra due momenti temporali diversi e in quali regioni del genoma, ovvero nelle regioni ORF, si sono verificati questi cambiamenti?
- Il virus si è evoluto attraverso inserimenti o delezioni nel suo genoma?
- Si è verificata una nuova introduzione di un virus non correlato nell'allevamento o al flusso?
- Sebbene sia meno probabile ma possibile, il nuovo virus ha acquisito delle modifiche nelle regioni genomiche prese di mira dalla RT-PCR o dal sequenziamento primer/sonda Sanger, impedendo ai test di rilevare il virus?
- Il virus ha subito una ricombinazione, cioè ha acquisito alcune regioni genomiche da due o più virus parentali?
La ricombinazione è un processo naturale dell'evoluzione del PRRSV e si verifica quando due PRRSV si replicano nella stessa cellula, generando un terzo virus derivato. La ricombinazione sarà approfondita in un Prossimo Articolo che pubblicheremo su 3tre3, intitolato "Le implicazioni del dilemma della ricombinazione del PRRSV".

